
Uncovering molecules with the desired attributes for developing novel drugs and materials is a slow and costly endeavor, utilizing extensive computational power and requiring many months of human effort to sift through the immense array of possible options.
Large language models (LLMs) such as ChatGPT might simplify this procedure, yet allowing an LLM to comprehend and draw conclusions regarding the atoms and bonds constituting a molecule, much like how it handles words forming sentences, poses a significant scientific challenge.
Scientists from MIT and the MIT-IBM Watson AI Lab have developed an innovative method that enhances a large language model using additional machine learning models called graph-based models. These specialized models are tailored for tasks involving the generation and prediction of molecular structures.
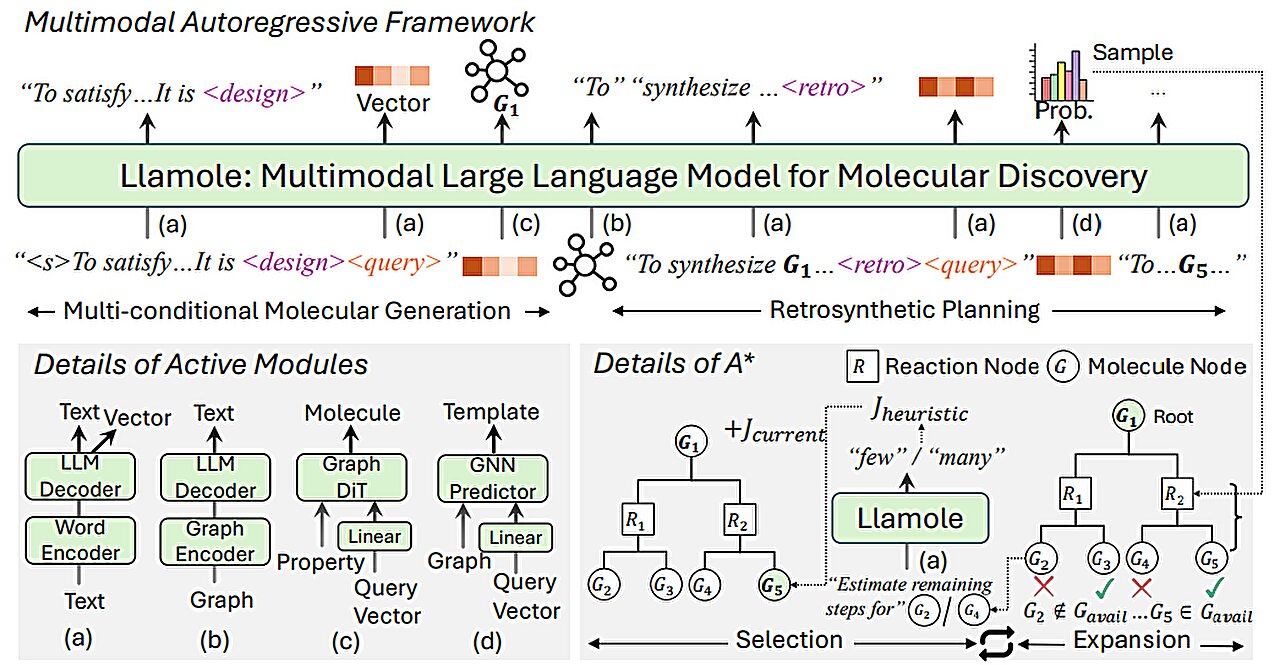
Their approach utilizes an underlying large language model (LLM) to understand natural-language inquiries about preferred molecular characteristics. The system dynamically alternates between using the primary LLM and employing specialized graph-based artificial intelligence components to craft the molecule, clarify the logic behind each decision, and outline a sequential procedure for synthesizing it. Throughout this process, it seamlessly integrates textual descriptions, graphical representations, and individual steps required for synthesis, merging these elements into a unified lexicon accessible to the LLM.
When compared to existing LLM-based approaches, this multimodal technique generated molecules that better matched user specifications and were more likely to have a valid synthesis plan, improving the success ratio from 5% to 35%.
It also outperformed LLMs that are more than 10 times its size and that design molecules and synthesis routes only with text-based representations, suggesting multimodality is key to the new system's success.
"This could hopefully be an end-to-end solution where, from start to finish, we would automate the entire process of designing and making a molecule. If an LLM could just give you the answer in a few seconds, it would be a huge time-saver for pharmaceutical companies," says Michael Sun, an MIT graduate student and co-author of a paper on this technique posted to the arXiv preprint server.
Sun's co-authors include lead author Gang Liu, a graduate student at the University of Notre Dame; Wojciech Matusik, a professor of electrical engineering and computer science at MIT who leads the Computational Design and Fabrication Group within the Computer Science and Artificial Intelligence Laboratory (CSAIL); Meng Jiang, associate professor at the University of Notre Dame; and senior author Jie Chen, a senior research scientist and manager in the MIT-IBM Watson AI Lab.
The study will be showcased at the International Conference on Learning Representations. ICLR 2025 ) taking place in Singapore from April 24 to 28.
Best of both worlds
Large language models aren't built to understand the nuances of chemistry, which is one reason they struggle with inverse molecular design, a process of identifying molecular structures that have certain functions or properties.
Large Language Models transform text into units known as tokens, which they utilize to consecutively forecast the subsequent word in a phrase. However, molecules are structured as graphs, made up of atoms and bonds without a specific sequence, posing challenges for their representation as linear text.
Conversely, potent graph-centric AI models depict atoms and molecular bonds as linked nodes and edges within a graphical structure. Although widely used for reverse molecular design, these models necessitate intricate input data, lack comprehension of natural language, and produce outcomes that may prove challenging to decipher.
The team from MIT merged an LLM with graph-based AI models into a cohesive framework designed to leverage the advantages of each approach.
Llamole, an acronym for Large Language Model for Molecular Discovery, utilizes a foundational LLM as a screening mechanism to comprehend a user’s inquiry—a straightforward request phrased in everyday language seeking a molecule with specific attributes.
For example, maybe someone is looking for a molecule capable of crossing the blood-brain barrier and blocking HIV, considering it needs to have a molecular weight of 209 along with specific bond properties.
When the large language model generates text in answer to the inquiry, it alternates among different graph modules.
A single component employs a graph diffusion mechanism to produce a molecular framework based on provided specifications. Another part utilizes a graph neural network to convert this created molecular configuration back into token form so that language models can process them. Lastly, another segment functions as a graph-based reaction forecaster; it accepts a preliminary molecular arrangement and forecasts a chemical transformation step-by-step, aiming to identify the precise series of actions needed to construct the compound starting with fundamental components.
The researchers developed a novel kind of trigger token designed to instruct the large language model (LLM) regarding when to engage each specific module. Upon detecting a "design" trigger token, the LLM transitions to the module responsible for drafting a molecular diagram; conversely, upon identifying a "retro" trigger token, it shifts to the module focused on retrochemical analysis, which forecasts the subsequent chemical synthesis stage.
"Essentially, all the output generated by the LLM prior to triggering a specific module is inputted into that module. This allows the module to learn operations that align with previous outputs," explains Sun.
In the same manner, the output of each module is encoded and fed back into the generation process of the LLM, so it understands what each module did and will continue predicting tokens based on those data.
Better, simpler molecular structures
Ultimately, Llamole generates an image depicting the molecular structure, a written explanation of the molecule, along with a detailed synthesis plan that includes all the steps required for its creation, right down to each specific chemical reaction involved.
In tests where molecules were designed according to specific user requirements, Llamole surpassed 10 conventional large language models (LLMs), four fine-tuned LLMs, as well as an advanced domain-specific technique. Additionally, it increased the success rate of retrosynthetic planning from 5% to 35%. This was achieved by producing molecules with superior quality—those featuring more straightforward designs and less expensive components for synthesis.
"Individually, large language models find it challenging to determine how to synthesize molecules due to the complexity involved in multi-step planning. Our approach can create more effective molecular structures that are simpler to produce," states Liu.
To train and evaluate Llamole, the researchers built two datasets from scratch since existing datasets of molecular structures didn't contain enough details. They augmented hundreds of thousands of patented molecules with AI-generated natural language descriptions and customized description templates.
The dataset used for refining the LLM encompasses templates linked to 10 specific molecular attributes; hence, one constraint of Llamole is that it is designed to create molecules based solely on these 10 quantitative characteristics.
For future research, the team aims to expand Llamole’s capabilities so it can handle various molecular properties. Additionally, they intend to enhance the graph modules to increase Llamole's effectiveness in retrosynthetic processes.
Ultimately, their aim is to extend this method to move past just molecules, developing multifunctional large language models capable of processing various kinds of graph-based information, like linked sensors within an electrical grid or exchanges in a stock market.
Chen states that Llamole showcases the potential of employing large language models as an interface for intricate data beyond mere textual descriptions. They foresee these models becoming a cornerstone that facilitates interaction with various AI algorithms to address all types of graph-related challenges.
More information: Gang Liu et al., Multimodal Large Language Models for Inverse Molecular Design Using Retrosynthetic Planning, arXiv (2024). DOI: 10.48550/arxiv.2410.04223
This story is republished courtesy of MIT News. web.mit.edu/newsoffice/ a well-known platform focusing on MIT’s latest research, innovations, and educational developments.
Furnished by Massachusetts Institute of Technology
This tale was initially released on Massima . Subscribe to our newsletter for the latest sci-tech news updates.